

PWS is caused by a lack of active genetic material in a particular region of chromosome 15 (15q11-q13). Normally, individuals inherit one copy of chromosome 15 from their mother and one from their father. The genes in the PWS region are normally only active on the chromosome that came from the father. In PWS, the genetic defect causing the inactivity of chromosome 15 from the father (paternal chromosome 15) can occur in one of three ways:
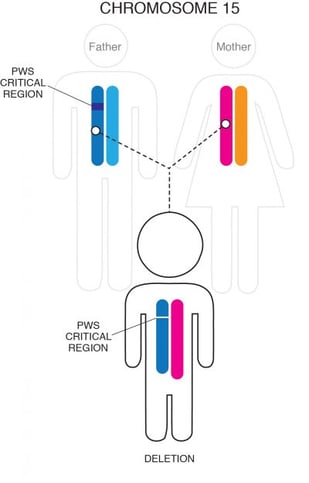
Most often, part of the chromosome 15 that was inherited from the person’s father is missing, or deleted, in this critical region. This small deletion occurs in approximately 60% of cases and usually is not detectable with routine genetic analysis such as amniocentesis.
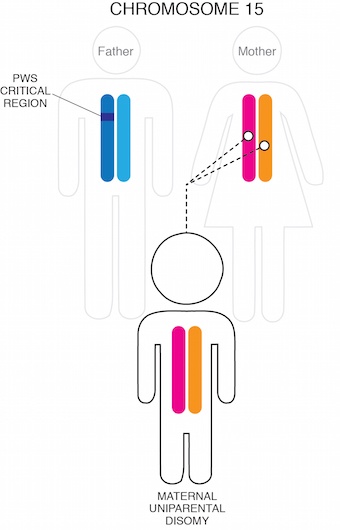
Another 35-40% of cases occur when an individual inherits two chromosome 15s from their mother and none from their father. This scenario is termed maternal uniparental disomy (UPD).
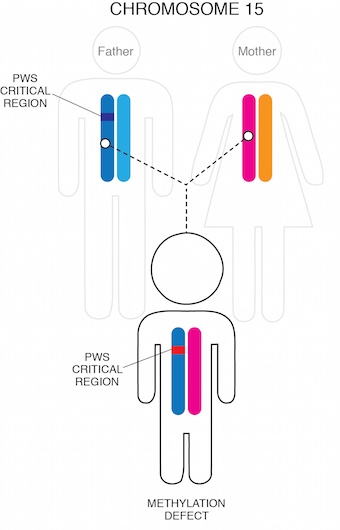
Finally, in a very small percentage of cases (1-3%), a small mutation in the Prader-Willi region causes the paternal chromosome 15 genetic material (although present) to be inactive.
The PWS region of chromosome 15 is one of the most complex regions of the human genome. Although there have been significant advances in understanding and characterizing the genetic changes associated with PWS, the exact mechanism by which lack of functional genetic material in this region leads to the symptoms associated with PWS is not understood. Scientists are actively studying the normal role of the genetic sequences in the PWS region and how their loss affects the hypothalamus and other systems in the body.
There may be some subtle differences in the characteristics of PWS based on genetic subtype: for example, those with deletions may be fair-skinned with light hair compared to other family members and may be more susceptible to seizures; those with PWS by UPD may be at higher risk for autism spectrum disorder and mental illness in young adulthood. Overall, however, there is considerable overlap between the different genetic subtypes. It is likely that the thousands of genes outside the PWS region, which exhibit normal variation between individuals, also contributes significantly to the variability in PWS symptoms between those with the disorder.
PWS is diagnosed with a blood test that looks for the genetic abnormalities that are specific to PWS – called a “methylation analysis.” A FISH (fluorescence in-situ hybridization) test identifies PWS by deletion, but it does not diagnose other forms of PWS. The methylation test will identify all types of PWS and is the preferred test for diagnosis. If a methylation test is done first, additional testing may be needed to determine whether PWS is caused by a paternal deletion, UPD, or an imprinting defect. In cases where an imprinting defect is suspected, blood may also be drawn from the parents.

Currently, there is no cure for PWS, and most research to date has been targeted towards treating specific symptoms (see Diagnosis & Treatments). For many individuals affected by the disorder, the elimination of some of the most difficult aspects of the syndrome, such as the insatiable appetite and obesity, would represent a significant improvement in quality of life and the ability to live independently.
The Foundation for Prader-Willi Research advances research that will lead to new treatments for PWS with the goal of an eventual cure. A number of clinical trials are underway to evaluate drugs for treating specific aspects of PWS, and FPWR is supporting studies exploring genetic therapy for PWS.



The Foundation for Prader-Willi Research (federal tax id 31-1763110) is a nonprofit corporation with federal tax exempt status as a public charity under section 501(c)(3).


![]()