


 Sometimes, valuable insights about “your” genetic disease can come from studies on entirely different genetic disorders that end up being connected in ways that weren’t easily appreciated at first glance.
Sometimes, valuable insights about “your” genetic disease can come from studies on entirely different genetic disorders that end up being connected in ways that weren’t easily appreciated at first glance.
Such is the case for a new study published by Drs. Ryan Potts and Christian Schaaf, who both have separate projects funded by FPWR, but who have worked collaboratively on this study. Dr. Schaaf recently showed that isolated MAGEL2 mutations cause a cluster of symptoms that are similar, but not identical to PWS (Schaaf, 2013, blogged here) You may recall that MAGEL2 is one of several genes in the PWS region of chromosome 15, and it is inactivated in PWS. Individuals with disruption of the MAGEL2 gene only typically show some features common to PWS - hypotonia at birth, developmental delay, hypogonadism, intellectual disability, and autism spectrum disorder. Other features of PWS, eg, increased appetite and obesity, are variable. Initially found in just a handful of patients, this disorder was referred to as a “PWS-like” syndrome, but the number of patients has grown and it has recently been given its own name, Schaaf-Yang syndrome.
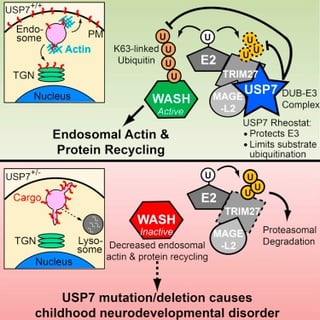
Dr. Potts has done some groundbreaking work (Hao, 2013) showing that the normal function of the MAGEL2 protein is one of general maintenance in the cell – it works as part of a specialized protein complex to direct the recycling of vesicles in the cell – an essential function that keeps receptors, signaling molecules, and other important proteins, contained in those vesicles, trafficked to the right places within the cell so they can do their jobs.
In a paper published today in Molecular Cell, Drs. Potts (UT Southwestern) and Schaaf (Baylor), along with coworkers at number of medical institutions, study a new disorder that shares features of Schaaf-Yang syndrome and PWS (USP7 Acts as a Molecular Rheostat to Promote WASH-Dependent Endosomal Protein Recycling and Is Mutated in a Human Neurodevelopmental Disorder). They find that another protein in the complex with MAGEL2, called USP7, also plays an important role in the vesicle recycling, acting to fine tune the process. Much of the paper focuses on carefully determining, at the molecular level, how the protein complex (which actually contains 3 protein: MAGEL2, TRIM27, and USP7) goes about regulating the vesicle recycling and trafficking. This provides a new understanding of this process, and is an important advance for general cell biology. Further, they highlight the critical function of this particular protein complex in neurons of the hypothalamus, a region of the brain thought to be dysfunctional in PWS. This suggests that a functional MAGEL2-TRIM27-USP7 complex may be critical for normal hypothalamic function.
Bringing the focus back to the clinic, a portion of the paper then looks to see if there are USP7 mutations associated with human genetic disorders. Searching through the DNA sequences of tens of thousands of individuals with unknown neurodevelopmental disorders, they found seven cases of mutations in USP7. Strikingly, the clinical characteristics of these individuals showed significant overlap when compared to Schaaf-Yang syndrome and PWS. USP7 mutations resulted in intellectual disability, autism spectrum disorder (at a higher frequency than typically seen in PWS, but similar to Schaaf-Yang), hypotonia, hypogonadism, aggressive behavior and an increased incidence of seizures. Thus, this study suggests that disruption of the MAGEL2-TRIM27-USP7 complex, through mutation of MAGEL2, mutation of USP7 or inactivation of MAGEL2 as in PWS, leads to similar clinical features. It also suggests a common target when considering the development of treatment for all of these disorders – restoring or replacing the normal function of the MAGEL2-TRIM27-USP7 complex might improve the clinical symptoms that are shared in individuals with USP7 mutations, Schaaf-Yang syndrome, and PWS.
A press release from Baylor College of Medicine, including an interesting twist on social media and rare diseases, can be found here.


