

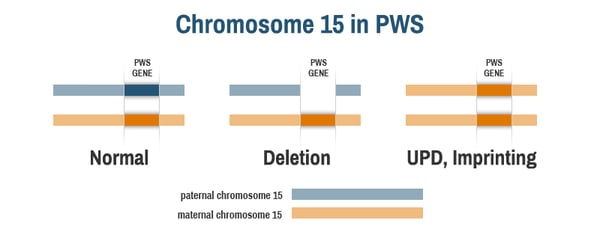
To understand how genetic therapy for PWS might work, it’s first important to know that the PWS region of chromosome 15 is imprinted, meaning that the genes behave differently depending on whether they were inherited from a person’s mother or father. In people without PWS, each cell has one copy of chromosome 15 inherited from their mother (the maternal chromosome, in orange below) and one copy of chromosome 15 inherited from their father (the paternal chromosome, in blue below). The PWS genes on chromosome 15 are only active on the paternal chromosome; they are inactive, or silent, on the maternal chromosome.
In people with PWS, the active, paternal copy of chromosome 15 is missing. Those with PWS by deletion have the full maternal chromosome 15, but the PWS genes are deleted on the paternal chromosome 15 (middle image). Those with PWS by uniparental disomy (UPD) or due to an imprinting defect have two copies of the maternal chromosome 15, but no paternal chromosome 15 (right image). Notably, all people with PWS have at least one copy of the maternally inherited chromosome 15. The PWS genes are present on this maternal chromosome, but they are inactive, or “silent.”
There are currently two general genetic therapy approaches being evaluated for the treatment of PWS:
Some of the possible strategies currently being investigated to achieve genetic therapy in PWS are described below, but importantly, an initial proof-of-concept study, funded by FPWR, has shown that genetic therapy might positively impact characteristics of PWS.
At this time, we do not know when the best time to introduce a potential gene therapy would be (e.g., infancy, childhood, or adulthood). Current research efforts include studies that will help scientists determine what benefit might be expected from doing gene therapy at different developmental stages.
Gene activation and gene replacement approaches for PWS would generally work similarly for PWS by deletion, UPD, and imprinting defects. However, there may be some differences in the effects of gene therapy in the different genetic subtypes. This is because there are two chromosome 15’s present in individuals with PWS by UPD or imprinting mutation, while there is only one set of PWS genes in individuals with PWS by deletion. Also, turning “on” some genes on chromosome 15 may result in other genes turning off. So “fine tuning” the level of PWS gene expression may be important, and there may be differences between the genetic subtypes. Studies in cells from individuals with PWS (deletion, UPD, and imprinting defect), and different mouse models of PWS will help scientists sort through the intricacies of how best to achieve genetic therapy in all subtypes of PWS.
We are fortunate to live in a time when gene therapy is progressing rapidly to the clinic for many genetic and acquired disorders. Lessons learned from genetic therapy research for Fragile X syndrome, Angelman syndrome, and other syndromes are helping smooth the path for genetic therapy for PWS, although of course each syndrome is unique and poses different challenges.





The Foundation for Prader-Willi Research (federal tax id 31-1763110) is a nonprofit corporation with federal tax exempt status as a public charity under section 501(c)(3).


![]()